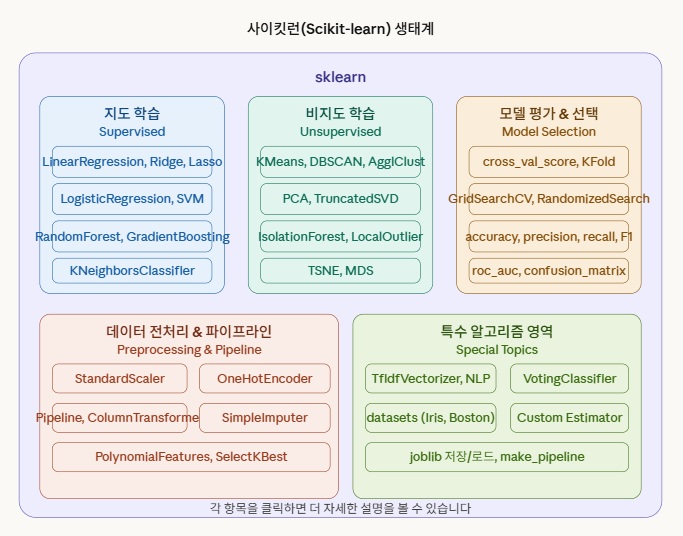
사이킷런이란?
2007년 구글 서머 오브 코드에서 시작된 오픈소스 프로젝트로, 현재 머신러닝 표준 라이브러리로 자리잡았습니다. NumPy, SciPy, Matplotlib 위에서 동작하며, 일관된 API 설계가 가장 큰 강점입니다.
핵심 개념: Estimator API
모든 알고리즘이 동일한 3가지 메서드로 통일되어 있습니다.
from sklearn.ensemble import RandomForestClassifier
model = RandomForestClassifier(n_estimators=100) # 1. 생성
model.fit(X_train, y_train) # 2. 학습
predictions = model.predict(X_test) # 3. 예측
전형적인 머신러닝 워크플로
from sklearn.model_selection import train_test_split, cross_val_score
from sklearn.preprocessing import StandardScaler
from sklearn.pipeline import Pipeline
from sklearn.svm import SVC
from sklearn.metrics import classification_report
데이터 분할
X_train, X_test, y_train, y_test = train_test_split(X, y, test_size=0.2)
파이프라인 구성 (전처리 + 모델을 하나로)
pipe = Pipeline([
(‘scaler’, StandardScaler()),
(‘model’, SVC(kernel=’rbf’))
])
교차 검증으로 모델 평가
scores = cross_val_score(pipe, X_train, y_train, cv=5)
print(f”CV 정확도: {scores.mean():.3f} ± {scores.std():.3f}”)
최종 학습 및 평가
pipe.fit(X_train, y_train)
print(classification_report(y_test, pipe.predict(X_test)))
주요 특징 정리
Pipeline이 특히 중요한데, 전처리와 모델을 하나의 객체로 묶어 데이터 누수(data leakage)를 방지하고 코드를 깔끔하게 유지해줍니다. 교차검증, 그리드서치도 Pipeline 단위로 적용됩니다.
fit() / transform() / predict() 세 메서드의 차이를 이해하는 게 핵심입니다. fit()은 학습 데이터로만, transform()은 학습된 파라미터를 적용, predict()는 최종 예측에 사용합니다.
RandomForestClassifier(n_estimators=100) 는 딱 두 가지를 하는 코드입니다 — “어떤 알고리즘을 쓸지” 고르고, “그 알고리즘의 설정값을 지정”하는 것이죠. 먼저 랜덤 포레스트가 무엇인지 시각적으로 보여드릴게요.
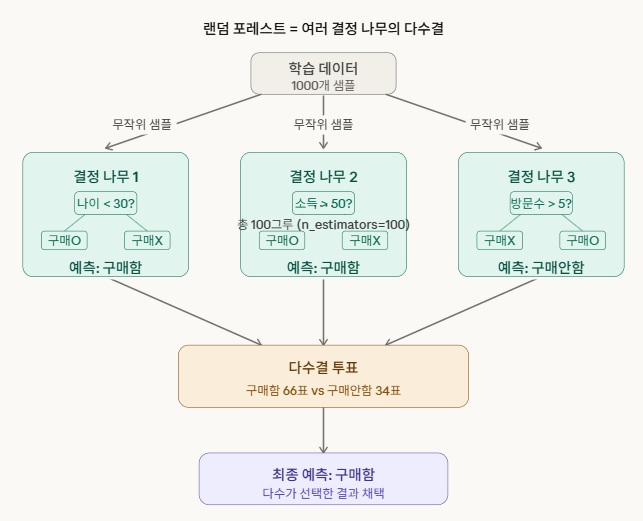
RandomForestClassifier(n_estimators=100) 해부하기
이 한 줄은 사실 두 부분으로 나뉩니다.
RandomForestClassifier — 어떤 알고리즘을 쓸지 선택하는 부분입니다. “나는 랜덤 포레스트 분류기를 사용하겠다”는 선언이에요. 랜덤 포레스트는 위 그림처럼 여러 결정 나무를 만들고 다수결로 예측하는 방식입니다.
n_estimators=100 — 그 알고리즘의 설정값(하이퍼파라미터)입니다. “나무를 몇 그루 만들 건지”를 지정해요. 100이면 100그루의 나무를 만들고 투표시킵니다.
n_estimators 숫자를 바꾸면 어떻게 될까?

자주 쓰는 다른 파라미터들
n_estimators 외에도 자주 설정하는 것들이 있습니다.
model = RandomForestClassifier(
n_estimators=100, # 나무 수 (기본값 100)
max_depth=5, # 나무 최대 깊이 (깊을수록 복잡)
min_samples_split=2, # 가지 나누는 최소 샘플 수
random_state=42 # 결과 재현을 위한 랜덤 시드
)
random_state=42는 특히 중요한데, 이걸 지정하지 않으면 실행할 때마다 나무 구성이 달라져서 결과가 바뀝니다. 연구나 팀 공유 시 같은 숫자를 써서 재현 가능하게 만드는 게 습관입니다.
모델 생성 공정
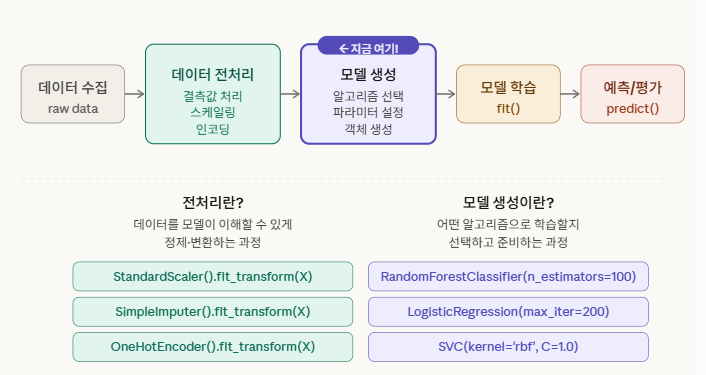
전처리 — “데이터를 손질하는 것” (재료 다듬기) 모델 생성 — “어떤 방법으로 학습할지 선택하는 것” (요리법 고르기)
요리에 비유하면, 채소를 씻고 썰고 양념하는 게 전처리고, RandomForestClassifier()는 “오늘은 볶음 요리로 하겠다”고 결정하는 것과 같습니다. 아직 불도 안 켠 상태예요. 실제로 불을 켜는 건 그 다음 단계인 model.fit()입니다.
그래서 전체 흐름을 코드로 보면 이렇게 됩니다.
① 전처리 단계
scaler = StandardScaler()
X_scaled = scaler.fit_transform(X) # 데이터를 숫자 크기 맞게 정규화
② 모델 생성 단계 ← 지금 배운 것
model = RandomForestClassifier(n_estimators=100) # 아직 아무것도 안 함
③ 학습 단계
model.fit(X_scaled, y) # 이때 비로소 나무 100그루를 키움
④ 예측 단계
predictions = model.predict(X_test)
RandomForestClassifier(n_estimators=100) 이 줄 자체는 사실 컴퓨터가 거의 아무 일도 안 합니다. 그냥 “설정지를 작성해서 서랍에 넣어두는” 것이고, 진짜 계산은 fit()을 호출할 때 시작됩니다.
“전처리된 데이터를 어떻게 학습시킬지 셋팅” — 이게 가장 정확한 표현입니다.
조금 더 구체적으로 말하면, 이 셋팅은 세 가지를 결정합니다.
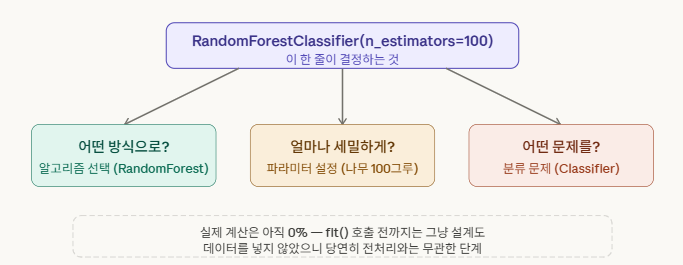
한 가지 재미있는 점은, 이 “셋팅” 단계에서는 데이터가 전혀 관여하지 않는다는 겁니다. 전처리된 데이터든 아니든 상관없이 먼저 모델을 만들어 둘 수 있어요. 마치 레시피를 정해두는 것과 같아서, 재료(데이터)가 준비되기 전에도 요리법(알고리즘)을 결정할 수 있는 거죠.
그래서 실제 코드에서도 이 순서가 엄격하지 않습니다.
이렇게 써도 되고
model = RandomForestClassifier(n_estimators=100) # 먼저 셋팅
X_scaled = scaler.fit_transform(X) # 나중에 전처리
model.fit(X_scaled, y)
이렇게 써도 똑같이 동작합니다
X_scaled = scaler.fit_transform(X) # 먼저 전처리
model = RandomForestClassifier(n_estimators=100) # 나중에 셋팅
model.fit(X_scaled, y)
경험칙 + 실험입니다. 데이터를 보지 않고 결정하는 게 아니라, 데이터의 특성을 사람이 먼저 파악한 다음 경험에 기반해 초기값을 정하는 거예요.
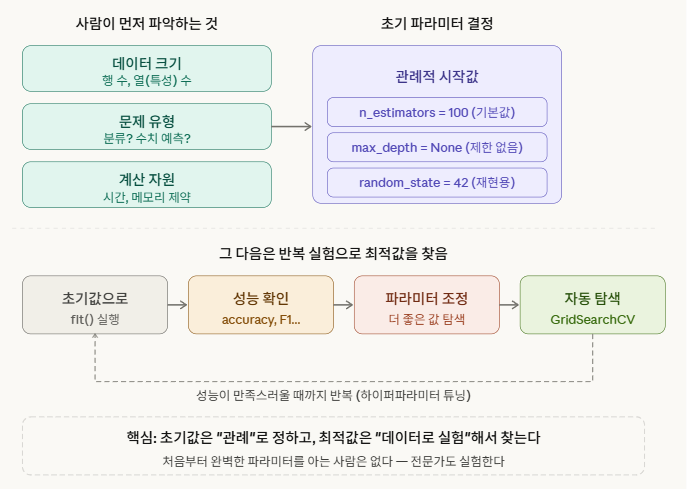
실제로 파라미터를 결정하는 근거는 세 가지 층위가 있습니다.
1단계 — 관례적 시작값 (경험칙) 수십 년간 수많은 사람들이 실험한 결과가 “기본값”으로 굳어진 겁니다. n_estimators=100은 “대부분의 데이터셋에서 무난하게 잘 작동하더라”는 커뮤니티 경험이 쌓인 값이에요. 그래서 사이킷런이 그걸 기본값으로 채택한 거고요.
2단계 — 데이터 특성에 따른 조정 데이터를 직접 보고 사람이 판단합니다.
데이터가 100만 행짜리 대용량이면 → 나무 적게
model = RandomForestClassifier(n_estimators=50)
데이터가 1000행짜리 소규모면 → 나무 넉넉히
model = RandomForestClassifier(n_estimators=300)
3단계 — GridSearchCV로 자동 탐색 (데이터 기반) 결국 최적값은 데이터로 실험해서 찾습니다. 이게 하이퍼파라미터 튜닝입니다.
from sklearn.model_selection import GridSearchCV
“이 범위에서 다 실험해봐” 라고 지시
param_grid = {
‘n_estimators’: [50, 100, 200, 300],
‘max_depth’: [3, 5, 10, None]
}
search = GridSearchCV(RandomForestClassifier(), param_grid, cv=5)
search.fit(X_train, y_train)
print(search.best_params_) # 실험 결과 최적값 출력
→ {‘max_depth’: 5, ‘n_estimators’: 200}
한 마디로 정리하면 — 처음엔 “경험으로 어림잡고”, 나중에 “데이터로 검증해서 다듬는” 방식입니다. 처음부터 완벽한 파라미터를 아는 전문가는 없고, 다들 이 과정을 거칩니다.
전처리 → 모델 셋팅(알고리즘+파라미터 결정) → fit()으로 학습 → 예측
파라미터는 처음엔 관례로 시작하고, 나중에 데이터로 실험해서 다듬는다 — 이 감각을 갖고 있으면 앞으로 다른 알고리즘을 만나도 같은 방식으로 접근할 수 있습니다.
사이킷 런에 들어 있는 알고리즘
많이 있습니다! 사이킷런은 사실 수십 가지 알고리즘을 제공해요. 크게 용도별로 나누면 이렇습니다.
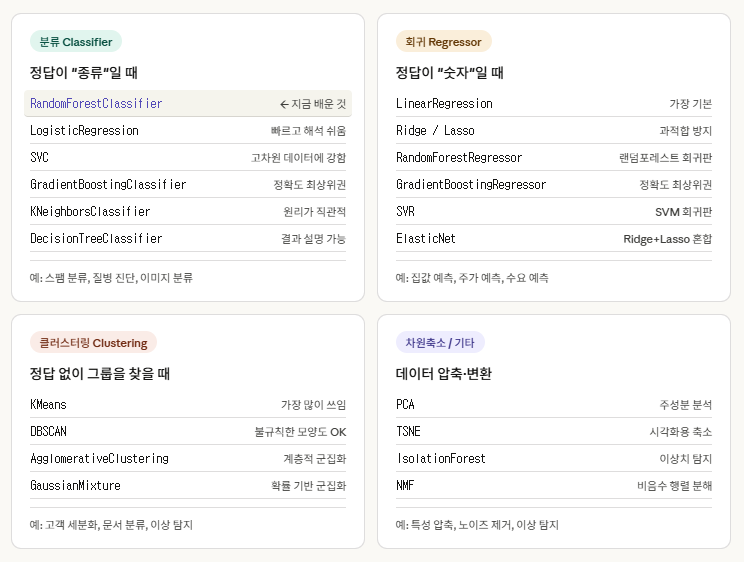
중요한 건 모든 모델이 같은 방식으로 사용된다는 점입니다.
랜덤포레스트
model = RandomForestClassifier(n_estimators=100)
로지스틱 회귀로 바꾸고 싶으면?
model = LogisticRegression(max_iter=200)
SVM으로 바꾸고 싶으면?
model = SVC(kernel=’rbf’)
어떤 모델이든 이후 코드는 똑같음
model.fit(X_train, y_train)
predictions = model.predict(X_test)
알고리즘이 바뀌어도 fit(), predict()는 그대로입니다. 사이킷런이 이 일관성을 설계 철학으로 삼은 덕분에, 모델을 바꾸는 게 딱 한 줄만 수정하면 되는 거예요.